Як зекономити 2 мільйони доларів і більше, або поговоримо про генетику

Валерій Зукін, віце-президент Української асоціації репродуктивної медицини, член правління Асоціації приватних медичних закладів України
Написати цю статтю мене спонукав шквал публікацій про пошук 2 мільйонів доларів на лікування приречених дітей зі спінальною м’язовою атрофією (СМА).
Чому саме зараз це рідкісне захворювання – спінальна м’язова атрофія – стало так часто згадуватися у ЗМІ? Чи раніше на нього не хворіли? Хворіли, і відоме воно з кінця 19 століття, коли австрійський лікар Гідо Вердніг і німецький – Йоганн Хоффманн описали це тяжке захворювання. Цікаво, що вже перші випадки хвороби були описані в одній родині, і це, без сумніву, наштовхувало лікарів на думку про генетичні причини захворювання. Але розвиток генетики кінця 19 століття явно не дозволяв робити якісь висновки.
Що ж це за хвороба – спінальна м’язова атрофія?
Я не збираюся докладно описувати це тяжке захворювання, його детальний опис є в Інтернеті. Якщо говорити коротко, за рахунок мутації у певному гені (якщо точно – SMN1) відбувається порушення мононейронів спинного мозку, і як наслідок – атрофія м’язів. Існує кілька форм цього захворювання. При найбільш тяжких формах дитина гине до 2-х років, при менш тяжких – виживає, але приречена бути інвалідом-візочником, який дуже часто потребує дихальної підтримки за допомогою апарата ШВЛ. Трагічним виглядає той факт, що в пацієнтів, які вижили, не страждає інтелект, і перелік відомих людей зі спінальною м’язовою атрофією є досить представницьким. А дитина, яка доживає до підліткового віку, виглядає ось так:

А до чого тут генетика?
Це захворювання належить до величезної групи захворювань з аутосомно-рецесивним успадкуванням. Існує великий каталог усіх описаних лікарями всього світу спадкових захворювань OMIM (Online Mendelian Inheritance in Man, omim.org). Там описано неймовірну кількість захворювань, й переважна їх більшість саме аутосомно-рецесивного типу успадкування. Давайте подивимось на схемі цей тип успадкування, що звучить так таємниче й інтригуюче:
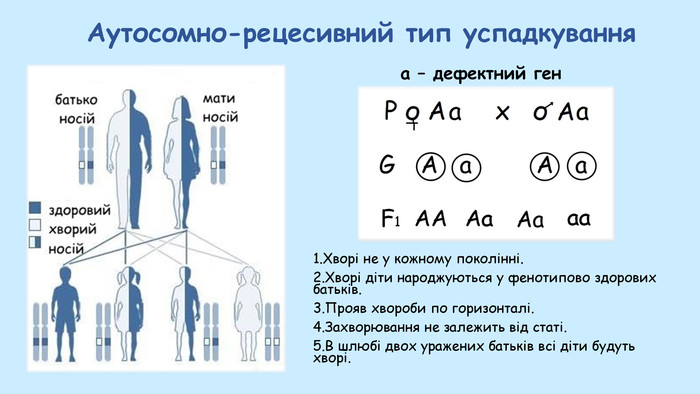
До речі, узята ця схема зі шкільного онлайн-курсу з біології для 10-го класу. Так-так, усі, хто принаймні має середню освіту, вивчали це у школі.
Які захворювання найбільш часто успадковуються за цим типом? Це вже згадувана спінальна м’язова атрофія, частота приблизно 1 на 6000 новонароджених. Але ще більш часте захворювання – муковісцидоз, частота – 1 випадок на 2500 новонароджених. Муковісцидоз – вкрай важка й невиліковна хвороба, 90 % пацієнтів не доживають до 2-х років.
Там само, у шкільному курсі біології, вчать і X-зчеплені захворювання, якими хворіють тільки чоловіки, а носіями мутантного гена є жінки.
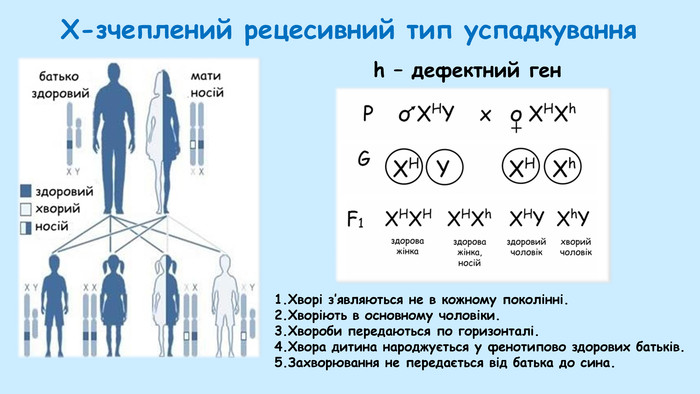
За цим типом успадковується всім відома гемофілія, схильність до незгортання крові, усім добре відома з історії царської родини Романових.
Але давайте повернемося до спінальної м’язової атрофії, яка зараз у всіх на вустах. Чому?
Лікування спінальної м’язової атрофії
Саме це захворювання стало одним із перших успішних випадків вдалого лікування. Спочатку у грудні 2016 року був дозволений до застосування в США препарат Спінраза. Після тривалих дискусій, він був дозволений до застосування в близько 40 країнах світу. Уводиться цей препарат у спинно-мозковий канал (люмбальна пункція), і лікування препаратом коштує 750 000 доларів у перший рік лікування, а потім 375 000 доларів щорічно пожиттєво. Попри таку дорожнечу, лікування Спінразою покривається державою в 40 країнах світу. Цей препарат належить до групи найдорожчих препаратів у світі. Тому його введення до програми покриття державою й супроводжувалося у багатьох країнах бурхливими дискусіями. Так, норвезький парламент назвав цей препарат “неетично дорогим” й спочатку відмовлявся покривати лікування СМА цим препаратом.
У травні 2019 року з’явився новий препарат для лікування СМА – Золгенсма. Не зупиняючись детально на складних механізмах його дії, згадаємо лише його головну відмінність від попередника – Спінрази. Золгенсма вводиться одноразово й внутрішньовенно. Усього одна ін’єкція коштує (увага, сядьте!) 2,1 мільйона доларів. Це не описка й не друкарська помилка. Саме така захмарна ціна дозволила цьому препарату посісти місце у Книзі рекордів Гіннеса як найдорожчому лікарському засобу.
Але питання перемоги над СМА – це не лише гроші. Які проблеми ще стоять перед пацієнтами, їх батьками й лікарями?
- Захворювання треба виявити якомога раніше. СМА може проявитися від народження, але є варіанти й більш пізнього проявлення цього підступного захворювання, у 6 – 12 місяців і пізніше. Тому зараз стоїть питання про включення до скринінгу (тотального обстеження) новонароджених і цього захворювання. Просто хочу нагадати, що в Україні всіх новонароджених обстежують (або мають обстежувати) на 4 захворювання. У світі цей список розширений до 40 захворювань. Включення СМА до цього списку робить ще більш дорожчим скринінг новонароджених. Щоб орієнтуватися, для України на найближчі десятиліття скринінг новонароджених на СМА й подальше лікування – малореальні.
- Найкращий ефект лікування за наявними даними досягається при початку лікування пацієнтів до 2-х місяців. Для Золгенсми термін початку лікування – 7 місяців. Якщо узагальнити наявні дані, то лікування можна починати і до 2-х років, але ефект буде скромніший. Дитина може почати сидіти, іноді стояти, зрідка – ходити з підтримкою. Для змучених батьків і це перемога, але абсолютно здоровою дитина так і не стане.
- Лікування цими препаратами не позбавлене певних ускладнень, іноді досить тяжких.
- І знову ж таки гроші, пресловуті гроші. Де їх узяти, якщо держава не може допомогти?
Що робити?
Єдиний ефективний механізм уникнути народження дитини хворої на спінальну м’язову атрофію – це обстеження всіх людей, які збираються обзавестися нащадками, на носійство гена SMN1. Для того, щоб порахувати ефективність цього підходу, необхідно мати знання в математиці й біології на рівні середньої школи.
Отже, носієм цього гена є 1 із 40 громадян України незалежно від статі. Ймовірність того, що зустрінуться 2 носії гена, – 1/40*1/40 = 1/1600. І при цій “зустрічі” ймовірність народження хворої дитини дорівнює 1/1600*1/4 = 1/6400. Чому ¼ ? А пригадайте вже згадувану програму із загальної біології для 10-го класу середньої школи. Й відразу все стане зрозумілим.
Знання – сила, – говорили древні. І не слід про це забувати. Озброєні силою знань ми легко порахуємо, що в Україні за 2020 рік народилося 293000 дітей. При частотності СМА 1 випадок на 6400 пологів мало народитися 46 (293000/6400) хворих дітей. Скільки їх народилося насправді, мені здається, не знає ніхто. Та й маленьким пацієнтам діагноз ще не поставлений.
Давайте пофантазуємо. Якщо обстежити всіх 293000 жінок, які народили в 2020 році, перед настанням вагітності або хоча б у ранні терміни (до 10 – 12 тижнів), то буде виявлено 7325 носіїв гена ( 293000/40 = 7325, де 40 – частотність носійства гена в нашій популяції). Наступним етапом необхідно обстежити їх чоловіків. Має бути виявлено 183 (7325/40 = 183) подружні пари з високим ризиком народження дитини зі СМА. Що їм робити?
У цих пар є такий вибір:
- провести передімплантаційну генетичну діагностику, а простіше – ЕКЗ з генетичним дослідженням ембріона;
- провести пренатальну діагностику, тобто дослідження плода під час вагітності, й не допустити народження хворої дитини.
Як показує вже наявний зарубіжний досвід, 70 - 80 % подружніх пар з високим ризиком обирають передімплантаційну діагностику. Інші – пренатальну діагностику або рідше інші способи запобігти народженню хворої дитини (аж до розірвання шлюбу чи відмови від дітонародження).
До речі, якщо нашу “віртуальну” програму профілактики СМА представити у фінансовому еквіваленті, то в масштабах України вона б коштувала приблизно 300 млн. гривень. Відповідне лікування неминуче народжених дітей – приблизно 3 млрд. гривень. Ураховуючи, що в нас немає ні профілактики, ні держава поки що не покриває лікування цих дітей, ці цифри існують зараз лише у віртуальній реальності.
Що далі?
Узагалі ж, те, що я розповів, відомо достатньо давно. Можливість виявити мутацію в гені СМА, а саме SMN1, появилася в 90-ті роки 20 століття. А в 2008 році обстеження на носійство цього гена було рекомендовано Американським Коледжем Акушерів і Гінекологів (The American College of Obstetricians and Gynecologists). Тоді рекомендували обстеження на носійство гена муковісцидозу. Ці два захворювання є найбільш частою причиною дитячої смертності від спадкових захворювань. Погляньмо на еволюцію скринінгу на носійство генетичних захворювань на такій схемі:

Скринінг на СМА і муковісцидоз з’явився у світі в середині 2000-х років, а з появою нових технологій, стало можливим одночасне дослідження на носійство від 100 до 400 генів. Мета гарна – не допустити народження хворої дитини! У США затребуваність скринінгових програм на носійство генів “оживила” політика страхових компаній. Логіка їхньої активності проста – виявилося, що набагато дешевше передбачити народження хворої дитини, ніж потім витрачати величезні гроші на її лікування.
А що в нас?
А нічого. За генетичними дослідженнями звертаються пацієнти, у яких вже “щось сталося”. Перспективних (профілактичних) досліджень вкрай мало. Чому? По-перше, не дуже наші майбутні батьки про це знають і думають. По-друге, не розвинутий в нас “страховий менталітет”. По-третє, якщо в родоводі все нормально, то не ймуть віри, що можуть потрапити в горезвісний “процент ризику”. Державних програм з профілактики спадкових захворювань у нас немає і в найближчому майбутньому не передбачається. Треба бути дуже великим оптимістом, щоб уявити, що мізерно фінансована українська медицина зможе забезпечити ВСІХ МАЙБУТНІХ БАТЬКІВ АБО ВАГІТНИХ на носійство генів. Тому спасіння потопаючих – справа рук самих потопаючих.
Чи можна в Україні пройти ці обстеження?
Можна. Проте витрати на маркетинг у загальноукраїнському масштабі вочевидь не під силу жодній приватній клініці чи лабораторії. А державної освітньої програми просто не існує. До речі, досвід багатьох країн демонструє, що проблема не тільки у фінансовому забезпеченні програм, але й в інформованості населення про життєву необхідность таких програм. Та й знання лікарів у галузі клінічної генетики в Україні, на жаль, далекі від ідеалу. Наша медична освіта питання медичної генетики явно проігнорувала.
PS. І все-таки я залишаюсь оптимістом. Безліч публікацій про збір коштів для лікування дітей, народжених зі СМА та іншими спадковими захворюваннями, так чи інакше продукує у суспільстві бажання не потратити до “сумної статистики”. І кожна родина, позбавлена від трагедії народження хворої дитини, і кожна ненароджена хвора дитина буде великою перемогою розуму людини над природою. До того ж це обстеження буде незрівнянно дешевшим, ніж 2 млн. доларів на лікування однієї дитини…
